Authors .Rabia B Choudry, MD Nestor Galvez-Jimenez, MD, MSc, MHSA, FACP Merit E Cudkowicz, MD, MSc Section Editors Jeremy M Shefner, MD, PhD Ira N Targoff, MD Deputy Editor John F Dashe, MD, PhD
Last literature review version 16.3: October 2008 | This topic last updated: May 13, 2008 (More)
INTRODUCTION — Amyotrophic lateral sclerosis (ALS), first described by Charcot in the nineteenth century [1], is a progressive neurodegenerative disorder that causes muscle weakness, disability, and eventually death, with a median survival of three to five years.
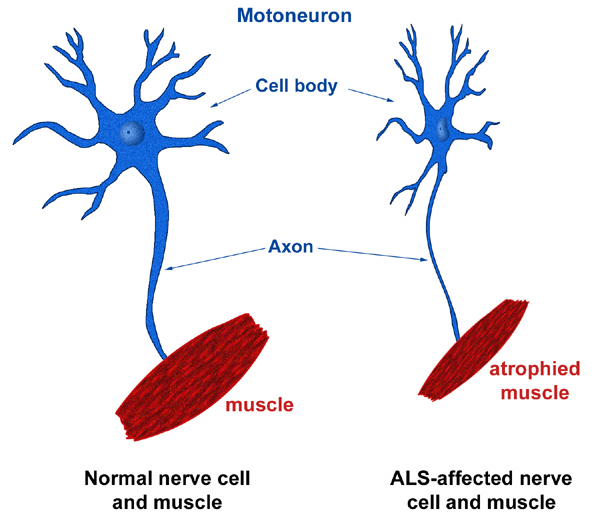
The hallmark of ALS is the combination of upper motor neuron (UMN) and lower motor neuron (LMN) involvement. The LMN findings of weakness, atrophy, and fasciculations are a direct consequence of muscle denervation. The UMN findings of hyperreflexia and spasticity result from degeneration of the lateral corticospinal tracts in the spinal cord [1].
The existing and experimental disease-modifying pharmacologic treatment of ALS will be reviewed here. The symptomatic management and the clinical features and diagnosis of ALS are discussed separately. (See "Symptom-based management of amyotrophic lateral sclerosis" and see "Clinical features of amyotrophic lateral sclerosis" and see "Diagnosis of amyotrophic lateral sclerosis").
DISEASE MODIFYING THERAPY — Amyotrophic lateral sclerosis (ALS) is a progressive neurologic disorder for which disease-specific treatment options are unsatisfactory. Of the currently available medications, only riluzole has been approved by the US Food and Drug Administration (FDA) for use in ALS.
Riluzole — Three separate mechanisms of riluzole are thought to reduce glutamate-induced excitotoxicity: inhibition of glutamic acid release, noncompetitive block of NMDA receptor mediated responses, and direct action on the voltage-dependent sodium channel [2]. Its precise mechanism of action in ALS is unclear [3].
Riluzole is the only drug to have any impact on survival in ALS. The evidence comes from two landmark clinical trials [4,5]:
• In a prospective, double-blind, placebo-controlled trial in 155 outpatients with ALS, survival at 12 months was significantly higher for patients receiving riluzole (100 mg/day) compared with controls (74 versus 58 percent, relative risk 0.43, CI 0.24-0.77) [4]. For the subset of patients with bulbar-onset ALS, an even greater advantage for survival at 12 months emerged for the riluzole group (73 versus 35 percent).
• In a larger follow-up study, 959 patients with clinically probable or definite ALS of less than five years duration were randomly assigned treatment with riluzole (50 mg, 100 mg, or 200 mg daily) or placebo [5]. After a median follow-up of 18 months, the primary outcome of survival without tracheostomy was significantly higher for the riluzole-treated group (100 mg/day) compared with controls (56.8 versus 50.4 percent, relative risk 0.57, CI 0.41-0.80).
Dose and side effects — The recommended dose of riluzole is 50 mg twice daily. It is well absorbed orally with a bioavailability of 60 percent and an elimination half-life of 12 hours. Metabolism is through the cytochrome P450 enzyme 1A2 (CYP1A2). The pharmacologic effects of riluzole may be affected by inhibitors of CYP1A2, such as theophylline and caffeine, which potentially may decrease the rate of riluzole elimination.
Riluzole is well tolerated, with the most significant adverse effects being gastrointestinal and hepatic. Neutropenia is extremely rare [6]. The most common adverse effects of riluzole are asthenia, dizziness, gastrointestinal disorders, and elevations in liver enzyme activities.
Elevation of the liver transaminases can be expected with riluzole treatment [7]. At least one alanine aminotransferase (ALT) level above the upper limit of normal (ULN) will occur in approximately half of patients treated with riluzole, while elevations greater than three or five times the ULN are seen in 8 and 2 percent of patients, respectively [8]. Liver function tests are indicated monthly for the first three months of riluzole treatment and every three months thereafter. (See "Riluzole: Drug information", section on Monitoring parameters).
Recommendations — The American Academy of Neurology has issued a practice advisory on the treatment of ALS with riluzole [9]. Patients most likely to benefit from treatment include those who have:
• Definite or probable ALS by El-Escorial criteria [10], in whom other causes of progressive muscle atrophy have been ruled out
• Symptoms present for less than five years
• Vital capacity (VC) greater than 60 percent of predicted
• No tracheostomy
Patients for whom no randomized data support the use of riluzole but expert opinion suggests potential benefit include those who have:
• Suspected or possible ALS by El-Escorial criteria
• Symptoms present for more than five years
• VC less than 60 percent of predicted
• Tracheostomy for prevention of aspiration only (ventilator independent)
Expert consensus suggests riluzole is of uncertain benefit in patients who have the following conditions:
• Tracheostomy required for ventilation
• Other incurable disorders
• Other forms of anterior horn cell disease
EXPERIMENTAL THERAPY — Multiple drugs have been tested for effectiveness in treating amyotrophic lateral sclerosis (ALS) since the advent ofriluzole as an FDA-approved treatment. While they have shown promise in preclinical in vitro and in vivo models of ALS, they have failed to show efficacy in human clinical trials. These include glutamate antagonists other than riluzole, neurotrophic factors, antioxidants, and immunomodulatory agents. The following illustrates the range of findings in some of these studies:
• Earlier trials found no benefit for verapamil [11], gabapentin [12], topiramate [13] or lamotrigine [14].
• A double-blind, placebo-controlled trial of celecoxib revealed no significant improvement in survival or beneficial effect on any of the outcome measures [15].
• In a randomized controlled trial with 412 patients, those assigned to minocycline (up to 400 mg daily) had significantly faster deterioration than those assigned to placebo, suggesting that minocycline treatment, or minocycline at the dose used in this trial, is harmful for patients with ALS [16].
• A phase II randomized controlled trial of 185 patients with ALS showed that the free radical scavenger coenzyme Q10, at up to 2700 mg daily, was safe [17]. However, the trial investigators concluded that coenzyme Q10 treatment was futile and should not be further tested as a possible treatment for ALS.
Animal models — Several animal models have been developed to investigate the pathogenesis and treatment of ALS. Earlier studies used pure motor neurons cultured in vitro [18]. In vitro models of cell death based on superoxide dismutase (SOD1) dysfunction were developed after the discovery of the Cu/Zn SOD1 gene abnormalities in familial ALS [19,20], and several mouse and rat models expressing mutant forms of SOD1 exist [21]. The experimentally induced mutations G93A, G37R, and G85R in the transgenic mouse models have phenotypes similar to human ALS [22]. There are also naturally occurring mouse models including the motor neuron degeneration (Mnd), progressive motor neuropathy (pmn) and wobbler.
The transgenic SOD1 mouse model is considered the most accurate representation of the disease process. However, the utility of this animal model for screening potential human ALS treatments is not established. A number of investigators have challenged its validity, since some experimental therapies that are effective in this model have not proven effective in human trials. Nevertheless, limitations in these trials related to dose range and sample size render direct comparison of animal to human studies quite speculative.
Insulin-like growth factor-I — Trials utilizing insulin-like growth factor-I (IGF-I) and neurotrophic factors have been unsuccessful to this point. The limitations and short-comings of these agents are related to unfavorable pharmacokinetics, bioavailability, and dose-limiting toxicities, in addition to the possibility that antibody inactivation may occur. The completed trials have shown that alternative methods of drug delivery, such as gene therapy may be warranted (see "Gene therapy" below).
Two preliminary human clinical trials employing recombinant IGF-I for ALS have reported somewhat discrepant results. The first studied IGF-I for nine months at doses of 0.05 mg/kg per day or 0.10 mg/kg per day [23]. No significant benefit was found for disease symptom progression, the primary outcome measure. However, treated groups showed a slower rate of both functional impairment progression and decline in health-related quality of life. In the second trial, subjects were randomized to receive either placebo or IGF-I 0.1 mg/kg per day subcutaneously for nine months [24]. No significant difference was found between treatment groups in terms of disease progression.
A third IGF-I trial funded by the National Institute of Neurological Disorders and Stroke (NINDS) is completed and results are expected in 2008 (NCT00035815, www.clinicaltrials.gov).
Antioxidants — Oxidative stress has been implicated in the pathogenesis of ALS due to the production of oxygen free radicals resulting in lipid peroxidation, cytoskeletal disruption, and damage to the mitochondria. However, trials utilizing antioxidants have been negative thus far in terms of delaying disease progression.
At least two randomized controlled trials have failed to demonstrate significant benefit of vitamin E as add-on therapy to riluzole in ALS. In the earlier study, subjects were randomly assigned to vitamin E 500 mg twice daily or placebo; there was no significant difference in disease progression at 12 months [25]. In a subsequent 18-month trial, subjects were randomly assigned to either megadose vitamin E at 5000 mg/day or placebo [26]. No significant difference was found between the rates of survival of the two groups. No significant adverse events were noted with dosages as high as 5000 mg per day.
In another randomized controlled trial testing the free radical scavenger N-acetylcysteine (NAC), there was no significant difference in delay of progression of the disease between the treatment and placebo groups [27]. However, there was a beneficial trend in survival for the patients with limb-onset disease.
Bioenergetic agents — In several transgenic mouse models of ALS with SOD1 mutations, early pathologic features include abnormalities in the mitochondria such as swelling and vacuolization [22]. The energy buffering agent creatine has been studied as a way to prevent or mitigate mitochondrial dysfunction. In transgenic mouse model data, oral administration of creatine showed dose-dependent benefit, with 1 percent creatine extending survival by 13 days and 2 percent creatine by 26 days [28]. This benefit seen with 2 percent creatine surpassed the extended survival seen with riluzole in this mouse model.
Despite the promise of creatine in animal modes, human randomized controlled trials have failed to demonstrate efficacy for creatine in the treatment of ALS. The first trial in the Netherlands evaluated 175 patients and found no significant benefit of creatine monohydrate 5 g twice daily for survival compared with placebo [29]. In a second trial, creatine monohydrate 20 mg daily for five days followed by 5 g daily showed no significant benefit compared with placebo in the delay of disease progression [30]. The doses of creatine used in these trials may have been too low to demonstrate effectiveness.
Antiapoptotic agents — A tricyclic selegiline analog, TCH346 (also called CGP3466) interacts with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in a mechanism that may prevent programmed cell death. It reduces the degeneration of motor neurons in many in vitro models of apoptosis [31], but it did not have significant effects on survival in transgenic mice. A 24-week phase human trial to evaluate four oral doses of TCH346 in over 500 subjects with ALS found that TCH346 did not slow the rate of disease progression or prolong survival at any of the doses tested [32].
Current and future trials — Despite the failures so far, a number of agents are under active investigation for the treatment of ALS including the following [33]:
• Arimoclomol
• Ceftriaxone
• Gene therapy
• Lithium
• R(+)pramipexole
Although some of these experimental therapeutics for ALS are available as prescription medications for other approved indications, or as over-the-counter medications, the off-label use of these drugs for the treatment of ALS is strongly discouraged. These drugs should be used to treat ALS only in the context of a clinical trial or under the discretion of an experienced physician.
A frequently updated list of drug trials can be found on the ALS Association website (www.alsa.org) and at www.clinicaltrials.gov.
Some agents currently in trial are described below.
Arimoclomol — Heat shock proteins are involved in protein repair, and thus are cytoprotective. Motor neurons appear to have a high threshold for activation of the heat shock protein pathway, and mutant SOD1 may contribute to reduced antiapoptotic capability [34]. Treatment of G93A mice with arimoclomol, a coinducer of heat shock proteins, delayed disease progression and improved survival by 22 percent [35]. A multicenter phase IIb study of arimoclomol in ALS is planned.
Ceftriaxone — Ceftriaxone and other beta lactam antibiotics appear to be active stimulators for expression of the glutamate transporter GLT1, also known as excitatory amino acid transporter type II (EEAT2) [36]. These compounds were identified using high throughput screening by the Neurodegeneration Drug Screening Consortium (NDSC) [36,37]. In vivo evaluation of ceftriaxone in normal rats also revealed upregulation of GLT1 protein expression, and transgenic G93A mice treated with ceftriaxone demonstrated a significant decline of motor neuron loss and hypercellular gliosis after two weeks of treatment [36].
A phase II/III human clinical trial to determine the safety and efficacy of long-term ceftriaxone treatment in subjects with ALS (NCT00349622,www.clinicaltrials.gov) is underway.
Gene therapy — Previous clinical trials with neurotrophic factors have shown equivocal to negative results (see "Insulin-like growth factor-I" above). Research in a transgenic mouse model has shown that IGF-I can be delivered directly to respiratory and motor limb muscles to target the affected motor neurons by using the retrograde transport ability of adeno-associated virus (AAV) [38]. IGF-I delivered by this vector delays disease onset and prolongs survival by 30 percent in the preclinical model and 18 percent in the postsymptomatic model in the SOD1 mutant mouse model.
In a similar method, glial cell line-derived neurotrophic factor (GDNF) gene was injected into lower extremity muscles of a G93A transgenic mouse using a replication defective adenoviral vector [39]. Larger motor neurons were seen in the injected muscles, suggesting gene therapy may delay progression in ALS.
Vascular endothelial growth factor (VEGF) is needed for angiogenesis and has been implicated in neuroprotection [40,41]. Researchers have shown that low levels of VEGF in transgenic mice can cause an ALS-like syndrome. Furthermore, intramuscular delivery of VEGF has shown delayed onset and prolonged survival in SOD1 mice.
In a study of intracerebroventricular delivery of VEGF gene therapy in transgenic rats, VEGF delayed disease onset, improved motor performance, and delayed the onset of paralysis [42]. This particular route was chosen over intrathecal delivery because of a speculated improved effect on bulbar and cervical motor neurons rather than motor neurons in the lumbar cord. VEGF gene therapy will likely be investigated in human trials in the near future.
Another approach to gene therapy employs antisense oligonucleotides to downregulate or silence mutant genes. The antisense strategy targets specific RNA sequences by constructing complementary oligonucleotides that bind to the native mRNA sequences and reduce their translation and subsequent protein expression.
In a preliminary study, continuous intraventricular infusion of antisense oligonucleotides to SOD1 reduced both SOD1 protein and mRNA levels throughout the rat brain and spinal cord [43]. In addition, this treatment significantly slowed disease progression when initiated near disease onset in a rat model of ALS caused by a SOD1 mutation.
Lithium — Lithium is an antiapoptotic agent that promotes autophagy [44,45], and may therefore have neuroprotective effects.
• Lithium prolongs survival in the G93A mouse model of ALS, with an increase in autophagic vacuoles noted in motor neurons [46,47].
• An open-label ALS clinical trial compared 16 patients treated with riluzole plus lithium to 28 patients treated with riluzole alone [47]. At the end of the 15 month follow-up period, all patients in the lithium group were alive, while mortality in the riluzole monotherapy group was 29 percent. In addition, disease progression, as measured by functional rating scales and pulmonary function, was slower in patients treated with lithium.
Several methodologic limitations of this clinical study make interpretation of the results very difficult, including lack of full blinding or proper randomization, small sample size, and inclusion of patients with slowly progressive ALS [47].
Based upon the findings reported in this study, double-blind randomized clinical trials are being planned. Until the results of such trials are available, lithium should NOT be used for off-label ALS treatment.
Use of UpToDate is subject to the Subscription and License Agreement.
REFERENCES
1. Rowland, LP. How amyotrophic lateral sclerosis got its name: the clinical-pathologic genius of Jean-Martin Charcot. Arch Neurol 2001; 58:512.
2. Riviere, M, Meininger, V, Zeisser, P, Munsat, T. An analysis of extended survival in patients with amyotrophic lateral sclerosis treated with riluzole. Arch Neurol 1998; 55:526.
3. Kennel, P, Revah, F, Bohme, GA, et al. Riluzole prolongs survival and delays muscle strength deterioration in mice with progressive motor neuronopathy (pmn). J Neurol Sci 2000; 180:55.
4. Bensimon, G, Lacomblez, L, Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994; 330:585.
5. Lacomblez, L, Bensimon, G, Leigh, P, et al. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Lancet 1996; 347:1425.
6. Weber, G, Bitterman, H. Riluzole-induced neutropenia. Neurology 2004; 62:1648.
7. Bensimon, G, Lacomblez, L, Delumeau, JC, et al. A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol 2002; 249:609.
8. Rilutek®(riluzole) tablets. Prescribing information as of September 2004a. Available online at http://products.sanofi-aventis.us/rilutek/rilutek.html. (Accessed 09/28/06).
9. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 1997; 49:657.
10. Brooks, BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of NeurologyResearch Group on Neuromuscular Diseases and the El Escorial "Clinical limits of amyotrophic lateral sclerosis" workshop contributors. J Neurol Sci 1994; 124 Suppl:96.
11. Miller, RG, Smith, SA, Murphy, JR, et al. A clinical trial of verapamil in amyotrophic lateral sclerosis. Muscle Nerve 1996; 19:511.
12. Miller, RG, Moore, DH, Gelinas, DF, et al. Phase III randomized trial of gabapentin in patients with amyotrophic lateral sclerosis. Neurology 2001; 56:843.
13. Cudkowicz, ME, Shefner, JM, Schoenfeld, DA, et al. A randomized, placebo-controlled trial of topiramate in amyotrophic lateral sclerosis. Neurology 2003; 61:456.
14. Ryberg, H, Askmark, H, Persson, L. A double-blind randomized clinical trial in amyotrophic lateral sclerosis using lamotrigine: effects on CSF glutamate, aspartate, branched-chain amino acid levels and clinical parameters. Acta Neurol Scand 2003; 108:1.
15. Cudkowicz, ME, Shefner, JM, Schoenfeld, DA, et al. Trial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol 2006; 60:22.
16. Gordon, PH, Moore, DH, Miller, RG, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol 2007; 6:1045.
17. Kaufmann, P, Thompson, JL, Levy, G, et al. Is a phase III trial of coenzyme Q10 (CoQ10) for ALS justified? Results of the phase II randomized controlled trial of CoQ10 for ALS. Presentation at the 60th annual meeting, American Academy of Neurology, Scientific Sessions, Boston, April 16, 2008.
18. Rothstein, JD, Kuncl, RW. Neuroprotective strategies in a model of chronic glutamate-mediated motor neuron toxicity. J Neurochem 1995; 65:643.
19. Pasinelli, P, Borchelt, DR, Houseweart, MK, et al. Caspase-1 is activated in neural cells and tissue with amyotrophic lateral sclerosis-associated mutations in copper-zinc superoxide dismutase. Proc Natl Acad Sci U S A 1998; 95:15763.
20. Pasinelli, P, Houseweart, MK, Brown, RH Jr, Cleveland, DW. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2000; 97:13901.
21. Flanagan, SW, Anderson, RD, Ross, MA, Oberley, LW. Overexpression of manganese superoxide dismutase attenuates neuronal death in human cells expressing mutant (G37R) Cu/Zn-superoxide dismutase. J Neurochem 2002; 81:170.
22. Wong, PC, Pardo, CA, Borchelt, DR, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995; 14:1105.
23. Lai, EC, Felice, KJ, Festoff, BW, et al. Effect of recombinant human insulin-like growth factor-I on progression of ALS. A placebo-controlled study. The North America ALS/IGF-I Study Group. Neurology 1997; 49:1621.
24. Borasio, GD, Robberecht, W, Leigh, PN, et al. A placebo-controlled trial of insulin-like growth factor-I in amyotrophic lateral sclerosis. European ALS/IGF-I Study Group. Neurology 1998; 51:583.
25. Desnuelle, C, Dib, M, Garrel, C, Favier, A. A double-blind, placebo-controlled randomized clinical trial of alpha-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole-tocopherol Study Group. Amyotroph Lateral Scler Other Motor Neuron Disord 2001; 2:9.
26. Graf, M, Ecker, D, Horowski, R, et al. High dose vitamin E therapy in amyotrophic lateral sclerosis as add-on therapy to riluzole: results of a placebo-controlled double-blind study. J Neural Transm 2005; 112:649.
27. Louwerse, ES, Weverling, GJ, Bossuyt, PM, et al. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch Neurol 1995; 52:559.
28. Klivenyi, P, Ferrante, RJ, Matthews, RT, et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med 1999; 5:347.
29. Groeneveld, GJ, Veldink, JH, van der, Tweel I, et al. A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Ann Neurol 2003; 53:437.
30. Shefner, J, Cudkowicz, M, Schoenfeld, D, et al. A Clinical Trial of Creatine in Amyotrophic Lateral Sclerosis. Neurology 2004; 63:1656.
31. Carlile, GW, Chalmers-Redman, RM, Tatton, NA, et al. Reduced apoptosis after nerve growth factor and serum withdrawal: conversion of tetrameric glyceraldehyde-3-phosphate dehydrogenase to a dimer. Mol Pharmacol 2000; 57:2.
32. Miller, R, Bradley, W, Cudkowicz, M, et al. Phase II/III randomized trialof TCH346 in patients with ALS. Neurology 2007; 69:776.
33. Traynor, BJ, Bruijn, L, Conwit, R, et al. Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology 2006; 67:20.
34. Batulan, Z, Shinder, GA, Minotti, S, et al. High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci 2003; 23:5789.
35. Kieran, D, Kalmar, B, Dick, JR, et al. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med 2004; 10:402.
36. Rothstein, JD, Patel, S, Regan, MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005; 433:73.
37. Heemskerk, J. High throughput drug screening. Amyotroph Lateral Scler Other Motor Neuron Disord 2004; 5 Suppl 1:19.
38. Kaspar, BK, Llado, J, Sherkat, N, et al. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 2003; 301:839.
39. Abe, K, Manabe, Y, Murakami, T. [Gene therapy and neurotrophic factor treatment for amyotrophic lateral sclerosis]. Rinsho Shinkeigaku 2001; 41:1160.
40. Jin, KL, Mao, XO, Greenberg, DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci U S A 2000; 97:10242.
41. Carmeliet, P. Blood vessels and nerves: common signals, pathways and diseases. Nat Rev Genet 2003; 4:710.
42. Storkebaum, E, Lambrechts, D, Dewerchin, M, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci 2005; 8:85.
43. Smith, RA, Miller, TM, Yamanaka, K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest 2006; 116:2290.
44. Sarkar, S, Floto, RA, Berger, Z, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170:1101.
45. Zhong, J, Yang, X, Yao, W, Lee, W. Lithium protects ethanol-induced neuronal apoptosis. Biochem Biophys Res Commun 2006; 350:905.
46. Shin, JH, Cho, SI, Lim, HR, et al. Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclerosis. Mol Pharmacol 2007; 71:965.
47. Fornai, F, Longone, P, Cafaro, L, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2008; 105:2052.